ProteinSequence#
- class biotite.sequence.ProteinSequence(sequence=())[source]#
Bases:
SequenceRepresentation of a protein sequence.
Furthermore this class offers a conversion of amino acids from 3-letter code into 1-letter code and vice versa.
- Parameters:
- sequenceiterable object, optional
The initial protein sequence. This may either be a list or a string. May take upper or lower case letters. If a list is given, the list elements can be 1-letter or 3-letter amino acid representations. By default the sequence is empty.
Notes
The
Alphabetof thisSequenceclass does not support selenocysteine. Please convert selenocysteine (U) into cysteine (C) or use a customSequenceclass, if the differentiation is necessary.- static convert_letter_1to3(symbol)#
Convert a 1-letter to a 3-letter amino acid representation.
- Parameters:
- symbolstring
1-letter amino acid representation.
- Returns:
- convertstring
3-letter amino acid representation.
- static convert_letter_3to1(symbol)#
Convert a 3-letter to a 1-letter amino acid representation.
- Parameters:
- symbolstring
3-letter amino acid representation.
- Returns:
- convertstring
1-letter amino acid representation.
- copy(new_seq_code=None)#
Copy the object.
- Parameters:
- new_seq_codendarray, optional
If this parameter is set, the sequence code is set to this value, rather than the original sequence code.
- Returns:
- copy
A copy of this object.
- static dtype(alphabet_size)#
Get the sequence code dtype required for the given size of the alphabet.
- get_alphabet()#
Get the
Alphabetof theSequence.This method must be overwritten, when subclassing
Sequence.- Returns:
- alphabetAlphabet
Sequencealphabet.
- get_molecular_weight(monoisotopic=False)#
Calculate the molecular weight of this protein.
Average protein molecular weight is calculated by the addition of average isotopic masses of the amino acids in the protein and the average isotopic mass of one water molecule.
- Parameters:
- monoisotopicbool
Use the mass of the most common isotope.
- Returns:
- weightfloat
Molecular weight of the protein represented by the sequence. Molecular weight values are given in Dalton (Da).
- get_symbol_frequency()#
Get the number of occurences of each symbol in the sequence.
If a symbol does not occur in the sequence, but it is in the alphabet, its number of occurences is 0.
- Returns:
- frequencydict
A dictionary containing the symbols as keys and the corresponding number of occurences in the sequence as values.
- is_valid()#
Check, if the sequence contains a valid sequence code.
A sequence code is valid, if at each sequence position the code is smaller than the size of the alphabet.
Invalid code means that the code cannot be decoded into symbols. Furthermore invalid code can lead to serious errors in alignments, since the substitution matrix is indexed with an invalid index.
- Returns:
- validbool
True, if the sequence is valid, false otherwise.
- remove_stops()#
Remove stop signals from the sequence.
- Returns:
- no_stopProteinSequence
A copy of this sequence without stop signals.
- reverse(copy=True)#
Reverse the
Sequence.- Parameters:
- copybool, optional
If set to False, the code
ndarrayof the returned sequence is an array view to the sequence code of this object. In this case, manipulations on the returned sequence would also affect this object. Otherwise, the sequence code is copied.
- Returns:
- reversedSequence
The reversed
Sequence.
Examples
>>> dna_seq = NucleotideSequence("ACGTA") >>> dna_seq_rev = dna_seq.reverse() >>> print(dna_seq_rev) ATGCA
Gallery#

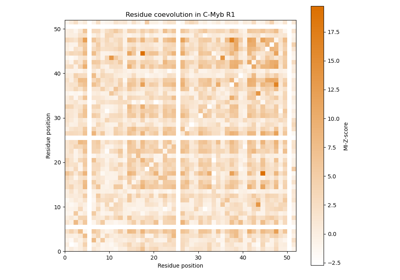
Mutual information as measure for coevolution of residues