biotite.application.sra.FastqDumpApp¶
- class biotite.application.sra.FastqDumpApp(uid, output_path_prefix=None, prefetch_path='prefetch', fasterq_dump_path='fasterq-dump', offset='Sanger')[source]¶
Bases:
_DumpAppFetch sequencing data from the NCBI sequence read archive (SRA) using sra-tools.
- Parameters
- uidstr
A unique identifier (UID) of the file to be downloaded.
- output_path_prefixstr, optional
The prefix of the path to store the downloaded FASTQ file.
.fastqis appended to this prefix if the run contains a single read per spot._1.fastq,_2.fastq, etc. is appended if it contains multiple reads per spot. By default, the files are created in a temporary directory and deleted after the files have been read.- prefetch_path, fasterq_dump_pathstr, optional
Path to the
prefetch_pathandfasterq-dumpbinary, respectively.- offsetint or {‘Sanger’, ‘Solexa’, ‘Illumina-1.3’, ‘Illumina-1.5’, ‘Illumina-1.8’}, optional
This value is subtracted from the FASTQ ASCII code to obtain the quality score. Can either be directly the value, or a string that indicates the score format.
- cancel()¶
Cancel the application when in RUNNING or FINISHED state.
- clean_up()¶
Do clean up work after the application terminates.
PROTECTED: Optionally override when inheriting.
- classmethod fetch(uid, output_path_prefix=None, prefetch_path='prefetch', fasterq_dump_path='fasterq-dump', offset='Sanger')¶
Get the sequences belonging to the UID from the NCBI sequence read archive (SRA).
- Parameters
- uidstr
A unique identifier (UID) of the file to be downloaded.
- output_path_prefixstr, optional
The prefix of the path to store the downloaded FASTQ file.
.fastqis appended to this prefix if the run contains a single read per spot._1.fastq,_2.fastq, etc. is appended if it contains multiple reads per spot. By default, the files are created in a temporary directory and deleted after the files have been read.- prefetch_path, fasterq_dump_pathstr, optional
Path to the
prefetch_pathandfasterq-dumpbinary, respectively.- offsetint or {‘Sanger’, ‘Solexa’, ‘Illumina-1.3’, ‘Illumina-1.5’, ‘Illumina-1.8’}, optional
This value is subtracted from the FASTQ ASCII code to obtain the quality score. Can either be directly the value, or a string that indicates the score format.
- Returns
- sequenceslist of dict (str -> NucleotideSequence)
This list contains the reads for each spot: The first item contains the first read for each spot, the second item contains the second read for each spot (if existing), etc. Each item in the list is a dictionary mapping identifiers to its corresponding sequence.
- get_app_state()¶
Get the current app state.
- Returns
- app_stateAppState
The current app state.
- get_fastq()¶
Get the FastqFile objects from the downloaded file(s).
- Returns
- fastq_fileslist of FastqFile
This list contains the reads for each spot: The first item contains the first read for each spot, the second item contains the second read for each spot (if existing), etc.
- get_fastq_dump_options()¶
Get additional options for the fasterq-dump call.
PROTECTED: Override when inheriting.
- Returns
- options: str
The additional options.
- get_file_paths()¶
Get the file paths to the downloaded files.
- Returns
- pathslist of str
The file paths to the downloaded files.
- get_prefetch_options()¶
Get additional options for the prefetch call.
PROTECTED: Override when inheriting.
- Returns
- options: str
The additional options.
- get_sequences()¶
Get the sequences from the downloaded file(s).
- Returns
- sequenceslist of dict (str -> NucleotideSequence)
This list contains the reads for each spot: The first item contains the first read for each spot, the second item contains the second read for each spot (if existing), etc. Each item in the list is a dictionary mapping identifiers to its corresponding sequence.
- get_sequences_and_scores()¶
Get the sequences and score values from the downloaded file(s).
- Returns
- sequences_and_scoreslist of dict (str -> (NucleotideSequence, ndarray))
This list contains the reads for each spot: The first item contains the first read for each spot, the second item contains the second read for each spot (if existing), etc. Each item in the list is a dictionary mapping identifiers to its corresponding sequence and score values.
- is_finished()¶
Check if the application has finished.
PROTECTED: Override when inheriting.
- Returns
- finishedbool
True of the application has finished, false otherwise
- join(timeout=None)¶
Conclude the application run and set its state to JOINED. This can only be done from the RUNNING or FINISHED state.
If the application is FINISHED the joining process happens immediately, if otherwise the application is RUNNING, this method waits until the application is FINISHED.
- Parameters
- timeoutfloat, optional
If this parameter is specified, the
Applicationonly waits for finishing until this value (in seconds) runs out. After this time is exceeded aTimeoutErroris raised and the application is cancelled.
- Raises
- TimeoutError
If the joining process exceeds the timeout value.
- start()¶
Start the application run and set its state to RUNNING. This can only be done from the CREATED state.
- wait_interval()¶
The time interval of
is_finished()calls in the joining process.PROTECTED: Override when inheriting.
- Returns
- intervalfloat
Time (in seconds) between calls of
is_finished()injoin()
Gallery¶
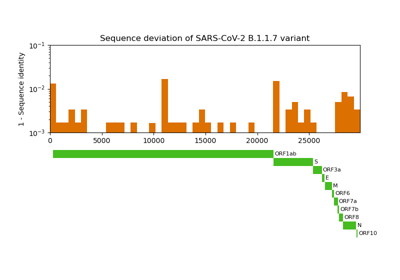
Comparative genome assembly of SARS-CoV-2 B.1.1.7 variant