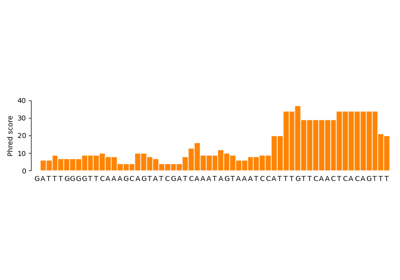
biotite.sequence.io.fastq.FastqFile¶
- class biotite.sequence.io.fastq.FastqFile(offset, chars_per_line=None)[source]¶
Bases:
TextFile,MutableMappingThis class represents a file in FASTQ format.
A FASTQ file stores one or multiple sequences (base calls) along with sequencing quality scores. Each sequence is associated with an identifer string, beginning with an
@.The quality scores are encoded as ASCII characters, with each actual score being the ASCII code subtracted by an offset value. The offset is format dependent. As the offset is not reliably deducible from the file contets, it must be provided explicitly, either as number or format (e.g.
'Illumina-1.8').Similar to the
FastaFileclass, this class implements theMutableMappinginterface: An identifier string (without the leading@) is used as index to get and set the corresponding sequence and quality.delremoves an entry in the file.- Parameters
- offsetint or {‘Sanger’, ‘Solexa’, ‘Illumina-1.3’, ‘Illumina-1.5’, ‘Illumina-1.8’}
This value is added to the quality score to obtain the ASCII code. Can either be directly the value, or a string that indicates the score format.
- chars_per_lineint, optional
The number characters in a line containing sequence data after which a line break is inserted. Only relevant, when adding sequences to a file. By default each sequence (and score string) is put into one line.
Examples
>>> import os.path >>> file = FastqFile(offset="Sanger") >>> file["seq1"] = str(NucleotideSequence("ATACT")), [0,3,10,7,12] >>> file["seq2"] = str(NucleotideSequence("TTGTAGG")), [15,13,24,21,28,38,35] >>> print(file) @seq1 ATACT + !$+(- @seq2 TTGTAGG + 0.96=GD >>> sequence, scores = file["seq1"] >>> print(sequence) ATACT >>> print(scores) [ 0 3 10 7 12] >>> del file["seq1"] >>> print(file) @seq2 TTGTAGG + 0.96=GD >>> file.write(os.path.join(path_to_directory, "test.fastq"))
- clear() None. Remove all items from D.¶
- copy()¶
Create a deep copy of this object.
- Returns
- copy
A copy of this object.
- get(k[, d]) D[k] if k in D, else d. d defaults to None.¶
- get_quality(identifier)¶
Get the quality scores for the specified identifier.
- Parameters
- identifierstr
The identifier of the quality scores.
- Returns
- scoresndarray, dtype=int
The quality scores corresponding to the identifier.
- get_seq_string(identifier)¶
Get the string representing the sequence for the specified identifier.
- Parameters
- identifierstr
The identifier of the sequence.
- Returns
- sequencestr
The sequence corresponding to the identifier.
- get_sequence(identifier)¶
Get the sequence for the specified identifier.
DEPRECATED: Use
get_seq_string()orget_sequence()instead.- Parameters
- identifierstr
The identifier of the sequence.
- Returns
- sequenceNucleotideSequence
The sequence corresponding to the identifier.
- items() a set-like object providing a view on D's items¶
- keys() a set-like object providing a view on D's keys¶
- pop(k[, d]) v, remove specified key and return the corresponding value.¶
If key is not found, d is returned if given, otherwise KeyError is raised.
- popitem() (k, v), remove and return some (key, value) pair¶
as a 2-tuple; but raise KeyError if D is empty.
- classmethod read(file, offset, chars_per_line=None)¶
Read a FASTQ file.
- Parameters
- filefile-like object or str
The file to be read. Alternatively a file path can be supplied.
- offsetint or {‘Sanger’, ‘Solexa’, ‘Illumina-1.3’, ‘Illumina-1.5’, ‘Illumina-1.8’}
This value is added to the quality score to obtain the ASCII code. Can either be directly the value, or a string that indicates the score format.
- chars_per_lineint, optional
The number characters in a line containing sequence data after which a line break is inserted. Only relevant, when adding sequences to a file. By default each sequence (and score string) is put into one line.
- Returns
- file_objectFastqFile
The parsed file.
- static read_iter(file, offset)¶
Create an iterator over each sequence (and corresponding scores) of the given FASTQ file.
- Parameters
- filefile-like object or str
The file to be read. Alternatively a file path can be supplied.
- offsetint or {‘Sanger’, ‘Solexa’, ‘Illumina-1.3’, ‘Illumina-1.5’, ‘Illumina-1.8’}
This value that is added to the quality score to obtain the ASCII code. Can either be directly the value, or a string that indicates the score format.
- Yields
- identifierstr
The identifier of the current sequence.
- sequencetuple(str, ndarray)
The current sequence as string and its corresponding quality scores as
ndarray.
Notes
This approach gives the same results as FastqFile.read(file, offset).items(), but is slightly faster and much more memory efficient.
- setdefault(k[, d]) D.get(k,d), also set D[k]=d if k not in D¶
- update([E, ]**F) None. Update D from mapping/iterable E and F.¶
If E present and has a .keys() method, does: for k in E: D[k] = E[k] If E present and lacks .keys() method, does: for (k, v) in E: D[k] = v In either case, this is followed by: for k, v in F.items(): D[k] = v
- values() an object providing a view on D's values¶
- write(file)¶
Write the contents of this object into a file (or file-like object).
- Parameters
- filefile-like object or str
The file to be written to. Alternatively a file path can be supplied.
- static write_iter(file, items, offset, chars_per_line=None)¶
Iterate over the given items and write each item into the specified file.
In contrast to
write(), the lines of text are not stored in an intermediateTextFile, but are directly written to the file. Hence, this static method may save a large amount of memory if a large file should be written, especially if the items are provided as generator.- Parameters
- filefile-like object or str
The file to be written to. Alternatively a file path can be supplied.
- itemsgenerator or array-like of tuple(str, tuple(str, ndarray))
The entries to be written into the file. Each entry consists of an identifier string and a tuple containing a sequence (as string) and a score array.
- offsetint or {‘Sanger’, ‘Solexa’, ‘Illumina-1.3’, ‘Illumina-1.5’, ‘Illumina-1.8’}
This value is added to the quality score to obtain the ASCII code. Can either be directly the value, or a string that indicates the score format.
- chars_per_lineint, optional
The number characters in a line containing sequence data after which a line break is inserted. Only relevant, when adding sequences to a file. By default each sequence (and score string) is put into one line.
Notes
This method does not test, whether the given identifiers are unambiguous.
Gallery¶
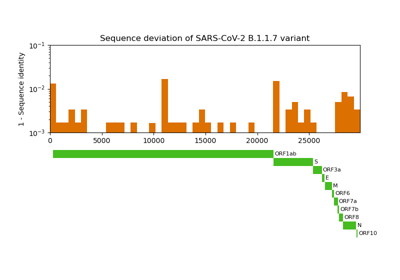
Comparative genome assembly of SARS-CoV-2 B.1.1.7 variant