


biotite.sequence.align.get_sequence_identity¶
- biotite.sequence.align.get_sequence_identity(alignment, mode='not_terminal')[source]¶
Calculate the sequence identity for an alignment.
The identity is equal to the matches divided by a measure for the length of the alignment that depends on the mode parameter.
- Parameters
- alignmentAlignment
The alignment to calculate the identity for.
- mode{‘all’, ‘not_terminal’, ‘shortest’}, optional
The calculation mode for alignment length.
all - The number of matches divided by the number of all alignment columns.
not_terminal - The number of matches divided by the number of alignment columns that are not terminal gaps in any of the sequences.
shortest - The number of matches divided by the length of the shortest sequence.
Default is not_terminal.
- Returns
- identityfloat
The sequence identity, ranging between 0 and 1.
See also
Gallery¶
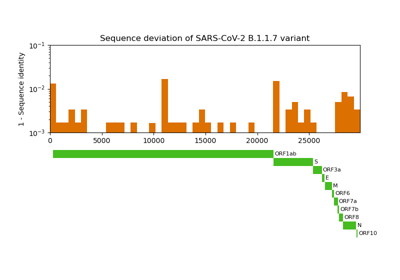
Comparative genome assembly of SARS-CoV-2 B.1.1.7 variant

Genome comparison between chloroplasts and cyanobacteria
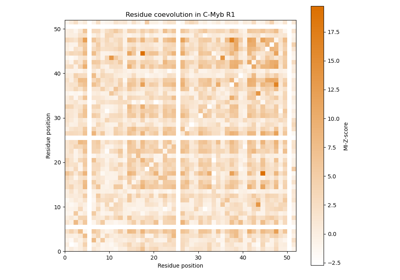
Mutual information as measure for coevolution of residues
