


biotite.sequence.align.SubstitutionMatrix¶
- class biotite.sequence.align.SubstitutionMatrix(alphabet1, alphabet2, score_matrix)[source]¶
Bases:
objectA
SubstitutionMatrixis the foundation for scoring in sequence alignments. ASubstitutionMatrixmaps each possible pairing of a symbol of a first alphabet with a symbol of a second alphabet to a score (integer).The class uses a 2-D (m x n)
ndarray(dtype=:attr:numpy.int32), where each element stores the score for a symbol pairing, indexed by the symbol codes of the respective symbols in an m-length alphabet 1 and an n-length alphabet 2.There are 3 ways to creates instances:
At first a 2-D
ndarraycontaining the scores can be directly provided.Secondly a dictionary can be provided, where the keys are pairing tuples and values are the corresponding scores. The pairing tuples consist of a symbol of alphabet 1 as first element and a symbol of alphabet 2 as second element. Parings have to be provided for each possible combination.
At last a valid matrix name can be given, which is loaded from the internal matrix database. The following matrices are avaliable:
- Nucleotide substitution matrices from NCBI database
NUC - Also usable with ambiguous alphabet
Protein substitution matrices from NCBI database
PAM<n>
BLOSUM<n>
MATCH - Only differentiates between match and mismatch
IDENTITY - Strongly penalizes mismatches
GONNET - Not usable with default protein alphabet
DAYHOFF
Corrected protein substitution matrices 1, <BLOCKS> is the BLOCKS version, the matrix is based on
BLOSUM<n>_<BLOCKS>
RBLOSUM<n>_<BLOCKS>
CorBLOSUM<n>_<BLOCKS>
A list of all available matrix names is returned by
list_db().Since this class can handle two different alphabets, it is possible to align two different types of sequences.
Objects of this class are immutable.
- Parameters
- alphabet1Alphabet, length=m
The first alphabet of the substitution matrix.
- alphabet2Alphabet, length=n
The second alphabet of the substitution matrix.
- score_matrixndarray, shape=(m,n) or dict or str
Either a symbol code indexed
ndarraycontaining the scores, or a dictionary mapping the symbol pairing to scores, or a string referencing a matrix in the internal database.
- Raises
- KeyError
If the matrix dictionary misses a symbol given in the alphabet.
References
- 1
M. Hess, F. Keul, M. Goesele, K. Hamacher, “Addressing inaccuracies in BLOSUM computation improves homology search performance,” BMC Bioinformatics, vol. 17, pp. 189, April 2016. doi: 10.1186/s12859-016-1060-3
Examples
Creating a matrix for two different (nonsense) alphabets via a matrix dictionary:
>>> alph1 = Alphabet(["foo","bar"]) >>> alph2 = Alphabet([1,2,3]) >>> matrix_dict = {("foo",1):5, ("foo",2):10, ("foo",3):15, ... ("bar",1):42, ("bar",2):42, ("bar",3):42} >>> matrix = SubstitutionMatrix(alph1, alph2, matrix_dict) >>> print(matrix.score_matrix()) [[ 5 10 15] [42 42 42]] >>> print(matrix.get_score("foo", 2)) 10 >>> print(matrix.get_score_by_code(0, 1)) 10
Creating an identity substitution matrix via the score matrix:
>>> alph = NucleotideSequence.alphabet_unamb >>> matrix = SubstitutionMatrix(alph, alph, np.identity(len(alph))) >>> print(matrix) A C G T A 1 0 0 0 C 0 1 0 0 G 0 0 1 0 T 0 0 0 1
Creating a matrix via database name:
>>> alph = ProteinSequence.alphabet >>> matrix = SubstitutionMatrix(alph, alph, "BLOSUM50")
- static dict_from_db(matrix_name)¶
Create a matrix dictionary from a valid matrix name in the internal matrix database.
The keys of the dictionary consist of tuples containing the aligned symbols and the values are the corresponding scores.
- Returns
- matrix_dictdict
A dictionary representing the substitution matrix.
- static dict_from_str(string)¶
Create a matrix dictionary from a string in NCBI matrix format.
Symbols of the first alphabet are taken from the left column, symbols of the second alphabet are taken from the top row.
The keys of the dictionary consist of tuples containing the aligned symbols and the values are the corresponding scores.
- Returns
- matrix_dictdict
A dictionary representing the substitution matrix.
- get_alphabet1()¶
Get the first alphabet.
- Returns
- alphabetAlphabet
The first alphabet.
- get_alphabet2()¶
Get the second alphabet.
- Returns
- alphabetAlphabet
The second alphabet.
- get_score(symbol1, symbol2)¶
Get the substitution score of two symbols.
- Parameters
- symbol1, symbol2object
Symbols to be aligned.
- Returns
- scoreint
The substitution / alignment score.
- get_score_by_code(code1, code2)¶
Get the substitution score of two symbols, represented by their code.
- Parameters
- code1, code2int
Symbol codes of the two symbols to be aligned.
- Returns
- scoreint
The substitution / alignment score.
- is_symmetric()¶
Check whether the substitution matrix is symmetric, i.e. both alphabets are identical and the score matrix is symmetric.
- Returns
- is_symmetricbool
True, if both alphabets are identical and the score matrix is symmetric, false otherwise.
- static list_db()¶
List all matrix names in the internal database.
- Returns
- db_listlist
List of matrix names in the internal database.
- score_matrix()¶
Get the 2-D
ndarraycontaining the score values.- Returns
- matrixndarray, shape=(m,n), dtype=np.int32
The symbol code indexed score matrix. The array is read-only.
- shape()¶
Get the shape (i.e. the length of both alphabets) of the subsitution matrix.
- Returns
- shapetuple
Matrix shape.
- static std_nucleotide_matrix()¶
Get the default
SubstitutionMatrixfor DNA sequence alignments.- Returns
- matrixSubstitutionMatrix
Default matrix.
- static std_protein_matrix()¶
Get the default
SubstitutionMatrixfor protein sequence alignments, which is BLOSUM62.- Returns
- matrixSubstitutionMatrix
Default matrix.
- transpose()¶
Get a copy of this instance, where the alphabets are interchanged.
- Returns
- transposedSubstitutionMatrix
The transposed substitution matrix.
Gallery¶

Pairwise sequence alignment of Avidin with Streptavidin

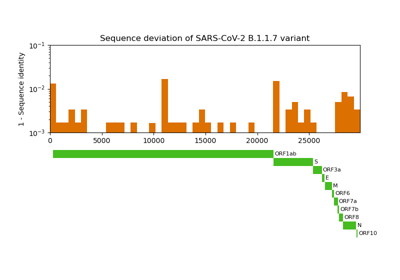
Comparative genome assembly of SARS-CoV-2 B.1.1.7 variant

Genome comparison between chloroplasts and cyanobacteria



Plot epitope mapping data onto protein sequence alignments

Structural alignment of lysozyme variants using ‘Protein Blocks’
