


Note
Go to the end to download the full example code
Structural alignment of lysozyme variants using ‘Protein Blocks’¶
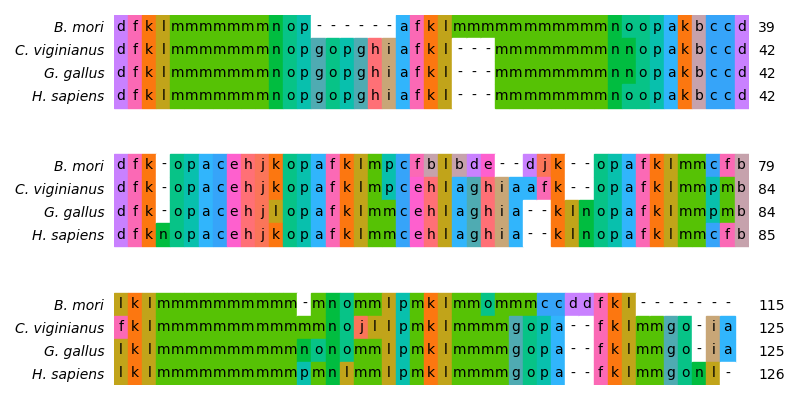
In this example we perform a structural alignment of multiple lysozyme variants from different organisms. A feasible approach to perfrom such a multiple structure alignment is the usage of a structural alphabet: At first the structure is translated into a sequence that represents the structure. Then the sequences can be aligned with the standard sequence alignment techniques, using the substitution matrix of the structural alphabet.
In this example, the structural alphabet we will use is called
protein blocks (PBs) 12:
There are 16 different PBs, represented by the symbols a to p.
Each one depicts a different set of the backbone dihedral angles of a
peptide 5-mer.
To assign a PB to an amino acid, the 5-mer centered on the respective
residue is taken, its backbone dihedral angles are calculated and the
PB with the least deviation to this set of angles is chosen.
- 1
A. G. de Brevern, C. Etchebest, S. Hazout, “Bayesian probabilistic approach for predicting backbone structures in terms of protein blocks,” Proteins: Structure, Function, and Bioinformatics, vol. 41, pp. 271–287, 2000. doi: 10.1002/1097-0134(20001115)41:3<271::AID-PROT10>3.0.CO;2-Z
- 2
J. Barnoud, H. Santuz, P. Craveur, A. P. Joseph, V. Jallu, A. G. de Brevern, P. Poulain, “PBxplore: a tool to analyze local protein structure and deformability with Protein Blocks,” PeerJ, vol. 5, pp. e4013, November 2017. doi: 10.7717/peerj.4013
# Code source: Patrick Kunzmann
# License: BSD 3 clause
from tempfile import gettempdir
import numpy as np
import matplotlib.pyplot as plt
import biotite.sequence as seq
import biotite.sequence.align as align
import biotite.sequence.graphics as graphics
import biotite.structure as struc
import biotite.structure.io.pdbx as pdbx
import biotite.database.rcsb as rcsb
# PB alphabet
pb_alphabet = seq.LetterAlphabet("abcdefghijklmnop")
# PB substitution matrix, adapted from PBxplore
matrix_str = """
a b c d e f g h i j k l m n o p
a 516 -59 113 -105 -411 -177 -27 -361 47 -103 -644 -259 -599 -372 -124 -83
b -59 541 -146 -210 -155 -310 -97 90 182 -128 -30 29 -745 -242 -165 22
c 113 -146 360 -14 -333 -240 49 -438 -269 -282 -688 -682 -608 -455 -147 6
d -105 -210 -14 221 5 -131 -349 -278 -253 -173 -585 -670 -1573 -1048 -691 -497
e -411 -155 -333 5 520 185 186 138 -378 -70 -112 -514 -1136 -469 -617 -632
f -177 -310 -240 -131 185 459 -99 -45 -445 83 -214 -88 -547 -629 -406 -552
g -27 -97 49 -349 186 -99 665 -99 -89 -118 -409 -138 -124 172 128 254
h -361 90 -438 -278 138 -45 -99 632 -205 316 192 -108 -712 -359 95 -399
i 47 182 -269 -253 -378 -445 -89 -205 696 186 8 15 -709 -269 -169 226
j -103 -128 -282 -173 -70 83 -118 316 186 768 196 5 -398 -340 -117 -104
k -644 -30 -688 -585 -112 -214 -409 192 8 196 568 -65 -270 -231 -471 -382
l -259 29 -682 -670 -514 -88 -138 -108 15 5 -65 533 -131 8 -11 -316
m -599 -745 -608 -1573 -1136 -547 -124 -712 -709 -398 -270 -131 241 -4 -190 -155
n -372 -242 -455 -1048 -469 -629 172 -359 -269 -340 -231 8 -4 703 88 146
o -124 -165 -147 -691 -617 -406 128 95 -169 -117 -471 -11 -190 88 716 58
p -83 22 6 -497 -632 -552 254 -399 226 -104 -382 -316 -155 146 58 609
"""
# PB reference angles, adapted from PBxplore
ref_angles = np.array([
[ 41.14, 75.53, 13.92, -99.80, 131.88, -96.27, 122.08, -99.68],
[108.24, -90.12, 119.54, -92.21, -18.06, -128.93, 147.04, -99.90],
[-11.61, -105.66, 94.81, -106.09, 133.56, -106.93, 135.97, -100.63],
[141.98, -112.79, 132.20, -114.79, 140.11, -111.05, 139.54, -103.16],
[133.25, -112.37, 137.64, -108.13, 133.00, -87.30, 120.54, 77.40],
[116.40, -105.53, 129.32, -96.68, 140.72, -74.19, -26.65, -94.51],
[ 0.40, -81.83, 4.91, -100.59, 85.50, -71.65, 130.78, 84.98],
[119.14, -102.58, 130.83, -67.91, 121.55, 76.25, -2.95, -90.88],
[130.68, -56.92, 119.26, 77.85, 10.42, -99.43, 141.40, -98.01],
[114.32, -121.47, 118.14, 82.88, -150.05, -83.81, 23.35, -85.82],
[117.16, -95.41, 140.40, -59.35, -29.23, -72.39, -25.08, -76.16],
[139.20, -55.96, -32.70, -68.51, -26.09, -74.44, -22.60, -71.74],
[-39.62, -64.73, -39.52, -65.54, -38.88, -66.89, -37.76, -70.19],
[-35.34, -65.03, -38.12, -66.34, -29.51, -89.10, -2.91, 77.90],
[-45.29, -67.44, -27.72, -87.27, 5.13, 77.49, 30.71, -93.23],
[-27.09, -86.14, 0.30, 59.85, 21.51, -96.30, 132.67, -92.91],
])
# Fetch animal lysoyzme structures
lyso_files = rcsb.fetch(
["1REX", "1AKI", "1DKJ", "1GD6"],
format="bcif", target_path=gettempdir()
)
organisms = ["H. sapiens", "G. gallus", "C. viginianus", "B. mori"]
# Create a PB sequence from each structure
pb_seqs = []
for file_name in lyso_files:
file = pdbx.BinaryCIFFile.read(file_name)
# Take only the first model into account
array = pdbx.get_structure(file, model=1)
# Remove everything but the first protein chain
array = array[struc.filter_amino_acids(array)]
array = array[array.chain_id == array.chain_id[0]]
# Calculate backbone dihedral angles,
# as the PBs are determined from them
phi, psi, omega = struc.dihedral_backbone(array)
# A PB requires the 8 phi/psi angles of 5 amino acids,
# centered on the amino acid to calculate the PB for
# Hence, the PBs are not defined for the two amino acids
# at each terminus
pb_angles = np.full((len(phi)-4, 8), np.nan)
pb_angles[:, 0] = psi[ : -4]
pb_angles[:, 1] = phi[1 : -3]
pb_angles[:, 2] = psi[1 : -3]
pb_angles[:, 3] = phi[2 : -2]
pb_angles[:, 4] = psi[2 : -2]
pb_angles[:, 5] = phi[3 : -1]
pb_angles[:, 6] = psi[3 : -1]
pb_angles[:, 7] = phi[4 : ]
pb_angles = np.rad2deg(pb_angles)
# Angle RMSD of all reference angles with all actual angles
rmsda = np.sum(
(
(
ref_angles[:, np.newaxis] - pb_angles[np.newaxis, :] + 180
) % 360 - 180
)**2,
axis=-1
)
# Chose PB, where the RMSDA to the reference angle is lowest
# Due to the definition of Biotite symbol codes
# the index of the chosen PB is directly the symbol code
pb_seq_code = np.argmin(rmsda, axis=0)
# Put the array of symbol codes into actual sequence objects
pb_sequence = seq.GeneralSequence(pb_alphabet)
pb_sequence.code = pb_seq_code
pb_seqs.append(pb_sequence)
# Perfrom a multiple sequence alignment of the PB sequences
matrix_dict = align.SubstitutionMatrix.dict_from_str(matrix_str)
matrix = align.SubstitutionMatrix(pb_alphabet, pb_alphabet, matrix_dict)
alignment, order, _, _ = align.align_multiple(
pb_seqs, matrix, gap_penalty=(-500,-100), terminal_penalty=False
)
# Visualize the alignment
# Order alignment according to guide tree
alignment = alignment[:, order.tolist()]
labels = [organisms[i] for i in order]
fig = plt.figure(figsize=(8.0, 4.0))
ax = fig.add_subplot(111)
# The color scheme was generated with the 'Gecos' software
graphics.plot_alignment_type_based(
ax, alignment, labels=labels, symbols_per_line=45, spacing=2,
show_numbers=True, color_scheme="flower"
)
# Organism names in italic
ax.set_yticklabels(ax.get_yticklabels(), fontdict={"fontstyle":"italic"})
fig.tight_layout()
plt.show()
