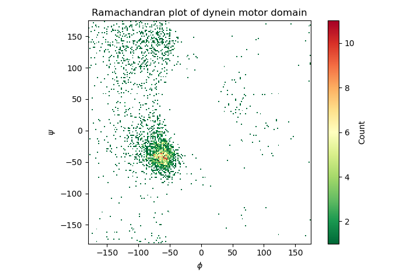
biotite.structure.dihedral_backbone¶
- biotite.structure.dihedral_backbone(atom_array)[source]¶
Measure the characteristic backbone dihedral angles of a protein structure.
- Parameters
- atom_array: AtomArray or AtomArrayStack
The protein structure. A complete backbone, without gaps, is required here. Chain transitions are allowed, the angles at the transition are NaN. The order of the backbone atoms for each residue must be (N, CA, C).
- Returns
- phi, psi, omegandarray
An array containing the 3 backbone dihedral angles for every CA. ‘phi’ is not defined at the N-terminus, ‘psi’ and ‘omega’ are not defined at the C-terminus. In these places the arrays have NaN values. If an
AtomArrayStackis given, the output angles are 2-dimensional, the first dimension corresponds to the model number.
- Raises
- BadStructureError
If the amount of backbone atoms is not equal to amount of residues times 3 (for N, CA and C).
See also
Examples
>>> phi, psi, omega = dihedral_backbone(atom_array) >>> print(np.stack([np.rad2deg(phi), np.rad2deg(psi)]).T) [[ nan -56.145] [ -43.980 -51.309] [ -66.466 -30.898] [ -65.219 -45.945] [ -64.747 -30.346] [ -73.136 -43.425] [ -64.882 -43.255] [ -59.509 -25.698] [ -77.989 -8.823] [ 110.784 8.079] [ 55.244 -124.371] [ -57.983 -28.766] [ -81.834 19.125] [-124.057 13.401] [ 67.931 25.218] [-143.952 131.297] [ -70.100 160.068] [ -69.484 145.669] [ -77.264 124.223] [ -78.100 nan]]
Gallery¶
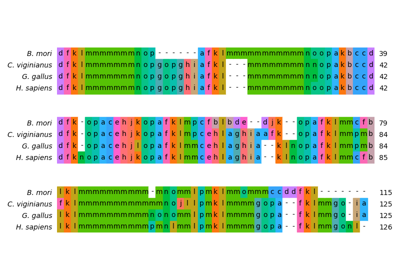
Structural alignment of lysozyme variants using ‘Protein Blocks’