


biotite.database.entrez.fetch¶
- biotite.database.entrez.fetch(uids, target_path, suffix, db_name, ret_type, ret_mode='text', overwrite=False, verbose=False)[source]¶
Download files from the NCBI Entrez database in various formats.
The data for each UID will be fetched into a separate file.
A list of valid database, retrieval type and mode combinations can be found under https://www.ncbi.nlm.nih.gov/books/NBK25499/table/chapter4.T._valid_values_of__retmode_and/?report=objectonly
This function requires an internet connection.
- Parameters
- uidsstr or iterable object of str
A single unique identifier (UID) or a list of UIDs of the file(s) to be downloaded.
- target_pathstr or None
The target directory of the downloaded files. If
None, the file content is stored in a file-like object (StringIO or BytesIO, respectively).- suffixstr
The file suffix of the downloaded files. This value is independent of the retrieval type.
- db_namestr:
E-utility or common database name.
- ret_typestr
Retrieval type
- ret_modestr, optional
Retrieval mode
- overwritebool, optional
If true, existing files will be overwritten. Otherwise the respective file will only be downloaded if the file does not exist yet in the specified target directory or if the file is empty. (Default: False)
- verbose: bool, optional
If true, the function will output the download progress. (Default: False)
- Returns
- filesstr or StringIO or BytesIO or list of (str or StringIO or BytesIO)
The file path(s) to the downloaded files. If a single string (a single UID) was given in uids, a single string is returned. If a list (or other iterable object) was given, a list of strings is returned. If target_path is
None, the file contents are stored in either StringIO or BytesIO objects.
Warning
Even if you give valid input to this function, in rare cases the database might return no or malformed data to you. In these cases the request should be retried. When the issue occurs repeatedly, the error is probably in your input.
See also
Examples
>>> import os.path >>> files = fetch(["1L2Y_A","3O5R_A"], path_to_directory, suffix="fa", ... db_name="protein", ret_type="fasta") >>> print([os.path.basename(file) for file in files]) ['1L2Y_A.fa', '3O5R_A.fa']
Gallery¶

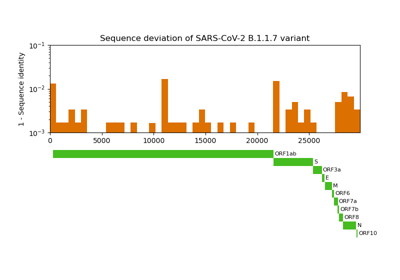
Comparative genome assembly of SARS-CoV-2 B.1.1.7 variant

Genome comparison between chloroplasts and cyanobacteria


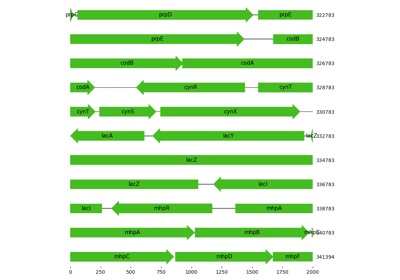
Visualization of region in proximity to lac operon
Three ways to get the secondary structure of a protein
