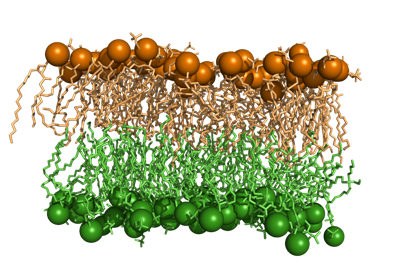
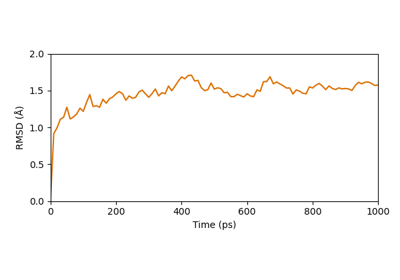
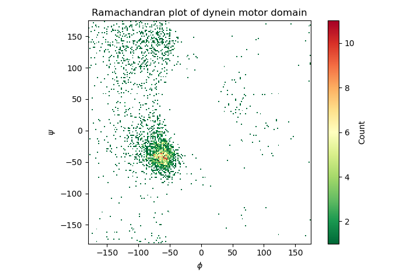
biotite.structure.io.load_structure¶
- biotite.structure.io.load_structure(file_path, template=None, **kwargs)[source]¶
Load an
AtomArrayor class`AtomArrayStack` from a structure file without the need to manually instantiate aFileobject.Internally this function uses a
Fileobject, based on the file extension. Trajectory files furthermore require specification of the template parameter.- Parameters
- file_pathstr
The path to structure file.
- templateAtomArray or AtomArrayStack or file-like object or str, optional
Only required when reading a trajectory file.
- kwargs
Additional parameters will be passed to either the
get_structure()orread()method of the file object. This does not affect files given via the template parameter. The only exception is the atom_i, which is applied to the template as well if number of atoms do not match.
- Returns
- arrayAtomArray or AtomArrayStack
If the file contains multiple models, an AtomArrayStack is returned, otherwise an AtomArray is returned.
- Raises
- ValueError
If the file format (i.e. the file extension) is unknown.
- TypeError
If a trajectory file is loaded without specifying the template parameter.
Gallery¶


Missing residues in protein structures of DNA-PKcs