


Note
Go to the end to download the full example code
Identification of lipid bilayer leaflets¶
This script implements the LeafletFinder algorithm 1 used by MDAnalysis. The algorithm detects which lipid molecules belong to the same membrane leaflet, i.e. the same side of a lipid bilayer, irrespective of the shape of the bilayer.
At first the algorithm creates an adjacency matrix of all lipid head groups, where the cutoff distance is smaller than the minimum distance between a head group of one leaflet to a head group of another leaflet. A graph is created from the matrix. Each leaflet is a connected subgraph.
- 1
N. {Michaud-Agrawal}, E. J. Denning, T. B. Woolf, O. Beckstein, “MDAnalysis: A toolkit for the analysis of molecular dynamics simulations,” Journal of Computational Chemistry, vol. 32, pp. 2319–2327, 2011. doi: 10.1002/jcc.21787
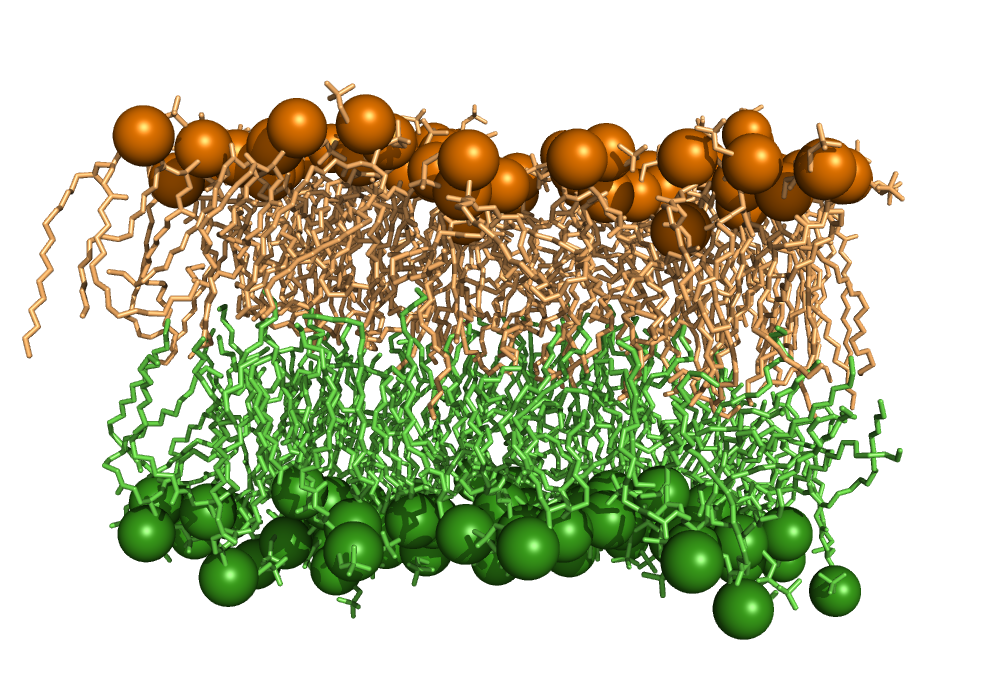
# Code source: Patrick Kunzmann
# License: BSD 3 clause
from tempfile import NamedTemporaryFile
import warnings
import numpy as np
import networkx as nx
import biotite.structure as struc
import biotite.structure.io as strucio
# The bilayer structure file can be downloaded from
# http://www.charmm-gui.org/archive/pure_bilayer/dppc.tar.gz
PDB_FILE_PATH = "../../download/dppc_n128.pdb"
def find_leaflets(structure, head_atom_mask,
cutoff_distance=15.0, periodic=False):
"""
Identify which lipids molecules belong to the same lipid bilayer
leaflet.
Parameters
----------
structure : AtomArray, shape=(n,)
The structure containing the membrane.
May also include other molecules, e.g. water or an embedded
protein.
head_atom_mask : ndarray, dtype=bool, shape=(n,)
A boolean mask that selects atoms from `structure` that
represent lipid head groups.
cutoff_distance : float, optional
When the distance of two head groups is larger than this value,
they are not (directly) connected in the same leaflet.
periodic : bool, optional,
If true, periodic boundary conditions are considered.
This requires that `structure` has an associated `box`.
Returns
-------
leaflets : ndarray, dtype=bool, shape=(m,n)
Multiple boolean masks, one for each identified leaflet.
Each masks indicates which atoms of the input `structure`
are in the leaflet.
"""
cell_list = struc.CellList(
structure, cell_size=cutoff_distance, selection=head_atom_mask,
periodic=periodic
)
adjacency_matrix = cell_list.create_adjacency_matrix(cutoff_distance)
graph = nx.Graph(adjacency_matrix)
head_leaflets = [sorted(c) for c in nx.connected_components(graph)
# A leaflet cannot consist of a single lipid
# This also removes all entries
# for atoms not in 'head_atom_mask'
if len(c) > 1]
# 'leaflets' contains indices to head atoms
# Broadcast each head atom index to all atoms in its corresponding
# residue
leaflet_masks = np.empty(
(len(head_leaflets), structure.array_length()),
dtype=bool
)
for i, head_leaflet in enumerate(head_leaflets):
leaflet_masks[i] = struc.get_residue_masks(structure, head_leaflet) \
.any(axis=0)
return leaflet_masks
# Suppress warning that elements were guessed,
# as this PDB file omits the 'chemical element' column
with warnings.catch_warnings():
warnings.simplefilter("ignore")
structure = strucio.load_structure(PDB_FILE_PATH)
# We cannot go over periodic boundaries in this case,
# because the input PDB does not define a box -> periodic=False
# However, as we have a planer lipid bilayer,
# periodicity should not matter
leaflets = find_leaflets(
structure,
head_atom_mask=(structure.res_name == "DPP") & (structure.atom_name == "P")
)
# Bilayer -> Expect two leaflets
assert len(leaflets) == 2
# Mark leaflets using different chain IDs
for chain_id, leaflet_mask in zip(("A", "B"), leaflets):
structure.chain_id[leaflet_mask] = chain_id
# Save marked lipids to structure file
temp = NamedTemporaryFile(suffix=".pdb")
strucio.save_structure(temp.name, structure)
# Visualization with PyMOL...
temp.close()
