


biotite.sequence.align.align_optimal¶
- biotite.sequence.align.align_optimal(seq1, seq2, matrix, gap_penalty=- 10, terminal_penalty=True, local=False, max_number=1000)[source]¶
Perform an optimal alignment of two sequences based on a dynamic programming algorithm.
This algorithm yields an optimal alignment, i.e. the sequences are aligned in the way that results in the highest similarity score. This operation can be very time and space consuming, because both scale linearly with each sequence length.
The aligned sequences do not need to be instances from the same
Sequencesubclass, since they do not need to have the same alphabet. The only requirement is that theSubstitutionMatrix’ alphabets extend the alphabets of the two sequences.This function can either perform a global alignment, based on the Needleman-Wunsch algorithm 1 or a local alignment, based on the Smith–Waterman algorithm 2.
Furthermore this function supports affine gap penalties using the Gotoh algorithm 3, however, this requires approximately 4 times the RAM space and execution time.
- Parameters
- seq1, seq2Sequence
The sequences to be aligned.
- matrixSubstitutionMatrix
The substitution matrix used for scoring.
- gap_penaltyint or tuple(int, int), optional
If an integer is provided, the value will be interpreted as linear gap penalty. If a tuple is provided, an affine gap penalty is used. The first integer in the tuple is the gap opening penalty, the second integer is the gap extension penalty. The values need to be negative. (Default: -10)
- terminal_penaltybool, optional
If true, gap penalties are applied to terminal gaps. If local is true, this parameter has no effect. (Default: True)
- localbool, optional
If false, a global alignment is performed, otherwise a local alignment is performed. (Default: False)
- max_numberint, optional
The maximum number of alignments returned. When the number of branches exceeds this value in the traceback step, no further branches are created. (Default: 1000)
- Returns
- alignmentslist, type=Alignment
A list of alignments. Each alignment in the list has the same maximum similarity score.
See also
References
- 1
S. B. Needleman, C. D. Wunsch, “A general method applicable to the search for similarities in the amino acid sequence of two proteins,” Journal of Molecular Biology, vol. 48, pp. 443–453, March 1970. doi: 10.1016/0022-2836(70)90057-4
- 2
T. F. Smith, M. S. Waterman, “Identification of common molecular subsequences,” Journal of Molecular Biology, vol. 147, pp. 195–197, March 1981. doi: 10.1016/0022-2836(81)90087-5
- 3
O. Gotoh, “An improved algorithm for matching biological sequences,” Journal of Molecular Biology, vol. 162, pp. 705–708, December 1982. doi: 10.1016/0022-2836(82)90398-9
Examples
>>> seq1 = NucleotideSequence("ATACGCTTGCT") >>> seq2 = NucleotideSequence("AGGCGCAGCT") >>> matrix = SubstitutionMatrix.std_nucleotide_matrix() >>> ali = align_optimal(seq1, seq2, matrix, gap_penalty=-6) >>> for a in ali: ... print(a, "\n") ATACGCTTGCT AGGCGCA-GCT ATACGCTTGCT AGGCGC-AGCT
Gallery¶

Pairwise sequence alignment of Avidin with Streptavidin
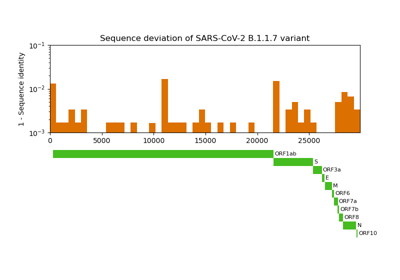
Comparative genome assembly of SARS-CoV-2 B.1.1.7 variant

Plot epitope mapping data onto protein sequence alignments
