


Note
Go to the end to download the full example code
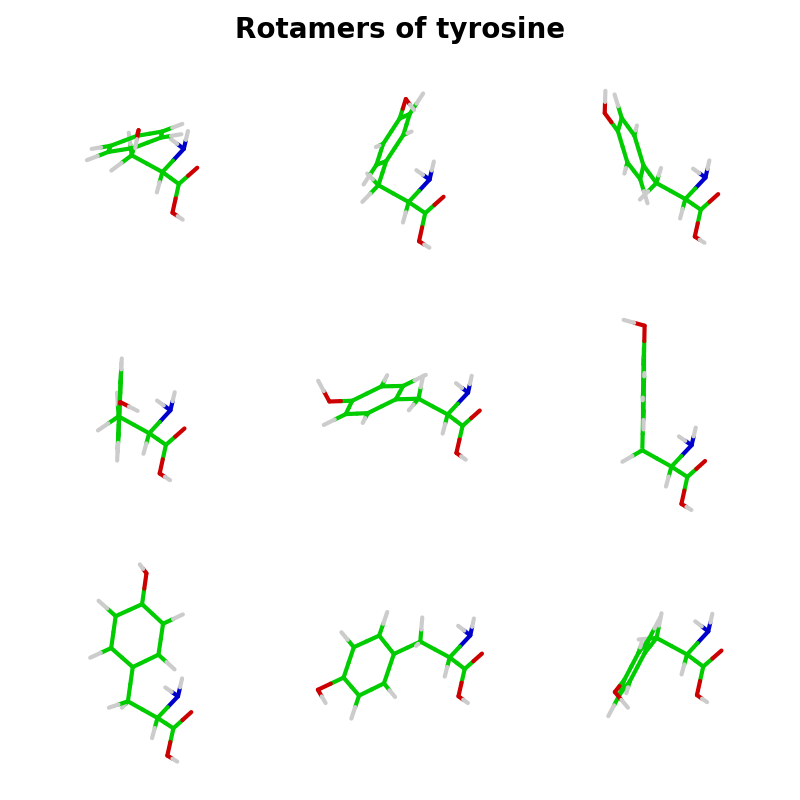
Creation of an amino acid rotamer library¶
This script creates rotamers for an amino acid, by randomly rotating about all rotatable bonds. In this case the rotamers are created for tyrosine.
Generally, this script could be used to sample possible conformations of an arbitrary small molecule.
Rotatable bonds in tyrosine:
CA <-> CB
CB <-> CG
CZ <-> OH
# Code source: Patrick Kunzmann
# License: BSD 3 clause
import numpy as np
import networkx as nx
import matplotlib.pyplot as plt
import biotite.structure as struc
import biotite.structure.io as strucio
import biotite.structure.info as info
import biotite.structure.graphics as graphics
# 'CA' is not in backbone,
# as we want to include the rotation between 'CA' and 'CB'
BACKBONE = ["N", "C", "O", "OXT"]
LIBRARY_SIZE = 9
# Get the structure (including bonds) from the standard RCSB compound
residue = info.residue("TYR")
bond_list = residue.bonds
### Identify rotatable bonds ###
rotatable_bonds = struc.find_rotatable_bonds(residue.bonds)
# Do not rotate about backbone bonds,
# as these are irrelevant for a amino rotamer library
for atom_name in BACKBONE:
index = np.where(residue.atom_name == atom_name)[0][0]
rotatable_bonds.remove_bonds_to(index)
print("Rotatable bonds in tyrosine:")
for atom_i, atom_j, _ in rotatable_bonds.as_array():
print(residue.atom_name[atom_i] + " <-> " + residue.atom_name[atom_j])
### VdW radii of each atom, required for the next step ###
vdw_radii = np.zeros(residue.array_length())
for i, element in enumerate(residue.element):
vdw_radii[i] = info.vdw_radius_single(element)
# The Minimum required distance between two atoms is mean of their
# VdW radii
vdw_radii_mean = (vdw_radii[:, np.newaxis] + vdw_radii[np.newaxis, :]) / 2
### Rotate randomly about bonds ###
np.random.seed(0)
rotamer_coord = np.zeros((LIBRARY_SIZE, residue.array_length(), 3))
for i in range(LIBRARY_SIZE):
# Coordinates for the current rotamer model
coord = residue.coord.copy()
for atom_i, atom_j, _ in rotatable_bonds.as_array():
# The bond axis
axis = coord[atom_j] - coord[atom_i]
# Position of one of the involved atoms
support = coord[atom_i]
# Only atoms at one side of the rotatable bond should be moved
# So the original Bondist is taken...
bond_list_without_axis = residue.bonds.copy()
# ...the rotatable bond is removed...
bond_list_without_axis.remove_bond(atom_i, atom_j)
# ...and these atoms are found by identifying the atoms that
# are still connected to one of the two atoms involved
rotated_atom_indices = struc.find_connected(
bond_list_without_axis, root=atom_i
)
accepted = False
while not accepted:
# A random angle between 0 and 360 degrees
angle = np.random.rand() * 2*np.pi
# Rotate
coord[rotated_atom_indices] = struc.rotate_about_axis(
coord[rotated_atom_indices], axis, angle, support
)
# Check if the atoms clash with each other:
# The distance between each pair of atoms must be larger
# than the sum of their VdW radii, if they are not bonded to
# each other
accepted = True
distances = struc.distance(
coord[:, np.newaxis], coord[np.newaxis, :]
)
clashed = distances < vdw_radii_mean
for clash_atom1, clash_atom2 in zip(*np.where(clashed)):
if clash_atom1 == clash_atom2:
# Ignore distance of an atom to itself
continue
if (clash_atom1, clash_atom2) not in bond_list:
# Nonbonded atoms clash
# -> structure is not accepted
accepted = False
rotamer_coord[i] = coord
rotamers = struc.from_template(residue, rotamer_coord)
### Superimpose backbone onto first model for better visualization ###
rotamers, _ = struc.superimpose(
rotamers[0], rotamers, atom_mask=struc.filter_peptide_backbone(rotamers)
)
### Visualize rotamers ###
colors = np.zeros((residue.array_length(), 3))
colors[residue.element == "H"] = (0.8, 0.8, 0.8) # gray
colors[residue.element == "C"] = (0.0, 0.8, 0.0) # green
colors[residue.element == "N"] = (0.0, 0.0, 0.8) # blue
colors[residue.element == "O"] = (0.8, 0.0, 0.0) # red
# For consistency, each subplot has the same box size
coord = rotamers.coord
size = np.array(
[coord[:, :, 0].max() - coord[:, :, 0].min(),
coord[:, :, 1].max() - coord[:, :, 1].min(),
coord[:, :, 2].max() - coord[:, :, 2].min()]
).max() * 0.5
fig = plt.figure(figsize=(8.0, 8.0))
fig.suptitle("Rotamers of tyrosine", fontsize=20, weight="bold")
for i, rotamer in enumerate(rotamers):
ax = fig.add_subplot(3, 3, i+1, projection="3d")
graphics.plot_atoms(ax, rotamer, colors, line_width=3, size=size, zoom=0.9)
fig.tight_layout()
plt.show()
### Write rotamers to structure file ###
#strucio.save_structure("rotamers.pdb", rotamers)
