


Note
Go to the end to download the full example code
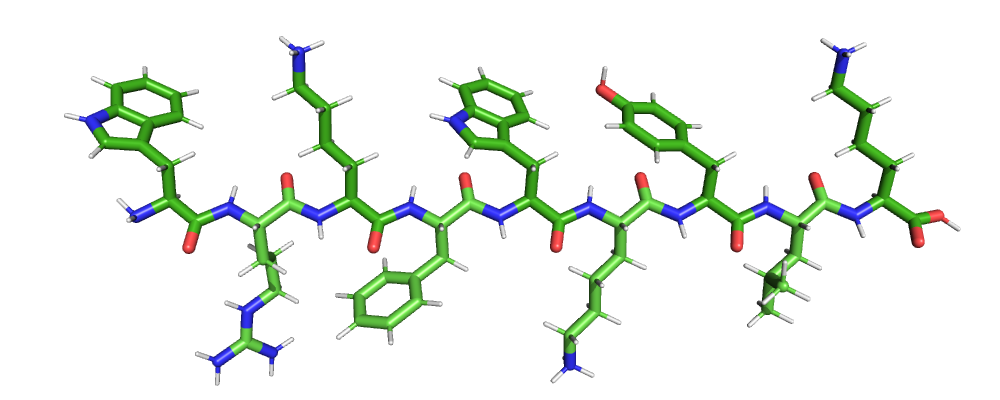
Assembly of a straight peptide from sequence¶
This script presents a function that takes an amino acid sequence and builds a straight peptide structure (like a \(\beta\)-strand) from it, including intramolecular bond information.
The function starts by building a backbone structure (N, CA, C) for all
residues in the sequence resulting in a ‘‘zigzag’’ chain.
Then for each amino acid, the respective side chain atoms and their
geometry are obtained from the reference PDB component dataset via
biotite.structure.info.residue() and are superimposed onto the
backbone chain.
The peptide bonds between the residues are formed and the atoms
lost in condensation are removed.
The geometry of the peptide oxygen and hydrogen atom is adjusted using
known peptide bond geometry taken from a reference structure.
# Code source: Patrick Kunzmann
# License: BSD 3 clause
from tempfile import NamedTemporaryFile
import itertools
import numpy as np
from numpy.linalg import norm
import biotite.sequence as seq
import biotite.structure as struc
import biotite.structure.io as strucio
import biotite.structure.info as info
C_N_LENGTH = 1.34
N_CA_LENGTH = 1.46
CA_C_LENGTH = 1.54
CA_C_N_ANGLE = 114
C_N_CA_ANGLE = 123
N_CA_C_ANGLE = 110
# Reference peptide bond atom coordinates taken from 1l2y:
# CA, C, N, O, H
peptide_coord = np.array([
[-8.608, 3.135, -1.618],
[-7.117, 2.964, -1.897],
[-6.379, 4.031, -2.228],
[-6.634, 1.849, -1.758],
[-6.821, 4.923, -2.394]
])
def create_raw_backbone_coord(number_of_res):
"""
Create coordinates for straight peptide chain in z-plane.
The peptide bonds are in trans configuration.
"""
coord = np.zeros((number_of_res * 3, 3))
for i, angle, angle_direction, length in zip(
range(len(coord)),
itertools.cycle([CA_C_N_ANGLE, C_N_CA_ANGLE, N_CA_C_ANGLE]),
itertools.cycle([1, -1]),
itertools.cycle([C_N_LENGTH, N_CA_LENGTH, CA_C_LENGTH])
):
if i == 0:
coord[i] = [0, 0, 0]
elif i == 1:
coord[i] = [0, length, 0]
else:
# Rotate about z-axis -> backbone lies in z-plane
rot_axis = [0, 0, angle_direction]
# Calculate the coordinates of a new atoms by rotating the previous
# bond by the given angle
new_coord = struc.rotate_about_axis(
coord[i-2],
axis = rot_axis,
angle = np.deg2rad(angle),
support = coord[i-1]
)
# Scale bond to correct bond length
bond_vector = new_coord - coord[i-1]
coord[i] = coord[i-1] + bond_vector * length / norm(bond_vector)
return coord
def append_residue(chain, residue):
"""
Append a residue to an existing chain.
Modify annotation arrays and remove atoms as necessary.
The atom coordinates are not altered.
"""
if chain.array_length() == 0:
# Chain is empty
residue.res_id[:] = 1
return residue
last_res_id = chain.res_id[-1]
# Remove atoms removed by peptide bond
chain = chain[
(chain.res_id != last_res_id) |
~np.isin(
chain.atom_name,
["OXT", "HXT"]
)
]
residue = residue[
~np.isin(
residue.atom_name,
["H2", "H3"]
)
]
# Increment residue ID for attached residue
residue.res_id[:] = last_res_id + 1
C_N_LENGTH = 1.34
N_CA_LENGTH = 1.46
CA_C_LENGTH = 1.54
CA_C_N_ANGLE = 114
C_N_CA_ANGLE = 123
N_CA_C_ANGLE = 110
# Reference peptide bond atom coordinates taken from 1l2y:
# CA, C, N, O, H
peptide_coord = np.array([
[-8.608, 3.135, -1.618],
[-7.117, 2.964, -1.897],
[-6.379, 4.031, -2.228],
[-6.634, 1.849, -1.758],
[-6.821, 4.923, -2.394]
])
def create_raw_backbone_coord(number_of_res):
"""
Create coordinates for straight peptide chain in z-plane.
The peptide bonds are in trans configuration.
"""
coord = np.zeros((number_of_res * 3, 3))
for i, angle, angle_direction, length in zip(
range(len(coord)),
itertools.cycle([CA_C_N_ANGLE, C_N_CA_ANGLE, N_CA_C_ANGLE]),
itertools.cycle([1, -1]),
itertools.cycle([C_N_LENGTH, N_CA_LENGTH, CA_C_LENGTH])
):
if i == 0:
coord[i] = [0, 0, 0]
elif i == 1:
coord[i] = [0, length, 0]
else:
# Rotate about z-axis -> backbone lies in z-plane
rot_axis = [0, 0, angle_direction]
# Calculate the coordinates of a new atoms by rotating the
# previous bond by the given angle
new_coord = struc.rotate_about_axis(
coord[i-2],
axis = rot_axis,
angle = np.deg2rad(angle),
support = coord[i-1]
)
# Scale bond to correct bond length
bond_vector = new_coord - coord[i-1]
coord[i] = coord[i-1] + bond_vector * length / norm(bond_vector)
return coord
def append_residue(chain, residue):
"""
Append a residue to an existing chain.
Modify annotation arrays and remove atoms as necessary.
The atom coordinates are not altered.
"""
if chain.array_length() == 0:
# Chain is empty
residue.res_id[:] = 1
return residue
last_res_id = chain.res_id[-1]
# Remove atoms removed by peptide bond
chain = chain[
(chain.res_id != last_res_id) |
~np.isin(
chain.atom_name,
["OXT", "HXT"]
)
]
residue = residue[
~np.isin(
residue.atom_name,
["H2", "H3"]
)
]
# Increment residue ID for attached residue
residue.res_id[:] = last_res_id + 1
# Append residue
chain += residue
# Add peptide bond
index_prev_c = np.where(chain.atom_name == "C")[0][-2]
index_curr_n = np.where(chain.atom_name == "N")[0][-1]
chain.bonds.add_bond(
index_prev_c, index_curr_n, struc.BondType.SINGLE
)
return chain
def assemble_peptide(sequence):
res_names = [seq.ProteinSequence.convert_letter_1to3(r) for r in sequence]
backbone_coord = create_raw_backbone_coord(len(sequence))
chain = struc.AtomArray(0)
for i, res_name in enumerate(res_names):
residue = info.residue(res_name)
# Superimpose residue to corresponding backbone coordinates
_, transformation = struc.superimpose(
backbone_coord[3*i : 3*i + 3],
residue.coord[np.isin(residue.atom_name, ["N", "CA", "C"])]
)
residue = transformation.apply(residue)
chain = append_residue(chain, residue)
if i != 0:
# Fix positions of peptide hydrogen and oxygen atom
ca_i, c_i, o_i = [
np.where(chain.atom_name == atom_name)[0][-2]
for atom_name in ["CA", "C", "O"]
]
n_i, h_i = [
np.where(chain.atom_name == atom_name)[0][-1]
for atom_name in ["N", "H"]
]
_, transformation = struc.superimpose(
chain.coord[[ca_i, c_i, n_i]],
peptide_coord[:3]
)
chain.coord[[o_i, h_i]] = transformation.apply(peptide_coord[3:])
return chain
# Sequence of an antimicrobial peptide
sequence = seq.ProteinSequence("WRKFWKYLK")
chain = assemble_peptide(sequence)
out_file = NamedTemporaryFile(suffix=".bcif", delete=False)
strucio.save_structure(out_file.name, chain)
# Visualization with PyMOL...
out_file.close()
