


biotite.sequence.align.Alignment¶
- class biotite.sequence.align.Alignment(sequences, trace, score=None)[source]¶
Bases:
objectAn
Alignmentobject stores information about which symbols of n sequences are aligned to each other and it stores the corresponding alignment score.Instead of saving a list of aligned symbols, this class saves the original n sequences, that were aligned, and a so called trace, which indicate the aligned symbols of these sequences. The trace is a (m x n)
ndarraywith alignment length m and sequence count n. Each element of the trace is the index in the corresponding sequence. A gap is represented by the value -1.Furthermore this class provides multiple utility functions for conversion into strings in order to make the alignment human readable.
Unless an
Alignmentobject is the result of an multiple sequence alignment, the object will contain only two sequences.All attributes of this class are publicly accessible.
- Parameters
- sequenceslist
A list of aligned sequences.
- tracendarray, dtype=int, shape=(n,m)
The alignment trace.
- scoreint, optional
Alignment score.
Examples
>>> seq1 = NucleotideSequence("CGTCAT") >>> seq2 = NucleotideSequence("TCATGC") >>> matrix = SubstitutionMatrix.std_nucleotide_matrix() >>> ali = align_optimal(seq1, seq2, matrix)[0] >>> print(ali) CGTCAT-- --TCATGC >>> print(ali.trace) [[ 0 -1] [ 1 -1] [ 2 0] [ 3 1] [ 4 2] [ 5 3] [-1 4] [-1 5]] >>> print(ali[1:4].trace) [[ 1 -1] [ 2 0] [ 3 1]] >>> print(ali[1:4, 0:1].trace) [[1] [2] [3]]
- Attributes
- sequenceslist
A list of aligned sequences.
- tracendarray, dtype=int, shape=(n,m)
The alignment trace.
- scoreint
Alignment score.
- get_gapped_sequences()¶
Get a the string representation of the gapped sequences.
- Returns
- sequenceslist of str
The list of gapped sequence strings. The order is the same as in Alignment.sequences.
- static trace_from_strings(seq_str_list)¶
Create a trace from strings that represent aligned sequences.
- Parameters
- seq_str_listlist of str
The strings, where each each one represents a sequence (with gaps) in an alignment. A
-is interpreted as gap.
- Returns
- tracendarray, dtype=int, shape=(n,2)
The created trace.
Gallery¶

Comparative genome assembly of SARS-CoV-2 B.1.1.7 variant

Genome comparison between chloroplasts and cyanobacteria


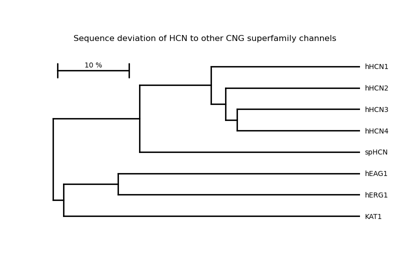


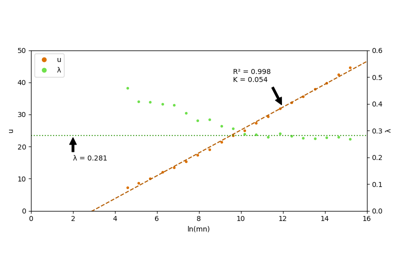

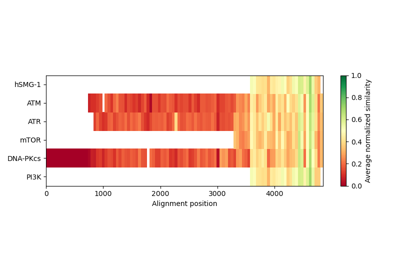

Plot epitope mapping data onto protein sequence alignments
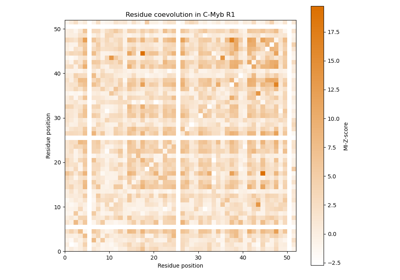
Mutual information as measure for coevolution of residues

Structural alignment of lysozyme variants using ‘Protein Blocks’
