


Note
Go to the end to download the full example code
BinaryCIF as trajectory format¶
This example demonstrates how the BinaryCIF format can be used as an alternative to classical trajectory formats (TRR, XTC, etc.).
For this purpose a trajectory file obtained from a MD simulation (Gromacs) of lysozyme (PDB: 1AKI) was loaded (101 frames, 50949 atoms), and the coordinates along with the frame number are put into a custom BinaryCIF category. For the model run length encoding is used. For the coordinates a combination of delta encoding and integer packing is used.
The trajectory file can be downloaded
here.
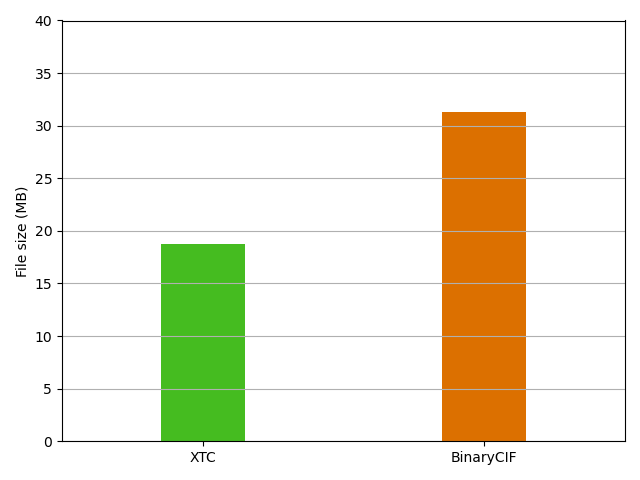
Using BinaryCIF for macromolecular trajectories takes advantage of the precise and open specification of the format and the wide support by a multitude of software. This comes at cost of a higher file size compared to XTC.
# Code source: Patrick Kunzmann
# License: BSD 3 clause
from tempfile import NamedTemporaryFile
import biotite
import biotite.structure.io.xtc as xtc
import biotite.structure.io.pdbx as pdbx
import numpy as np
import matplotlib.pyplot as plt
import os.path
# Put here the path of the downloaded trajectory file
xtc_file_path = "../../download/lysozyme_md.xtc"
xtc_file = xtc.XTCFile.read(xtc_file_path)
coord = xtc_file.get_coord()
n_frames = coord.shape[0]
n_atoms = coord.shape[1]
# [1, 1, ..., 1, 1, 2, 2, ..., 2, 2, n, n, ..., n, n] for n frames
frames = np.repeat(np.arange(1, n_atoms + 1), n_frames)
columns = {}
columns["frame"] = pdbx.BinaryCIFData(
frames,
encoding=[
pdbx.RunLengthEncoding(src_type=np.int32),
pdbx.ByteArrayEncoding(),
],
)
for i, dim in enumerate(("x", "y", "z")):
columns[f"coord_{dim}"] = pdbx.BinaryCIFData(
coord[:,:,i].flatten(),
encoding=[
pdbx.FixedPointEncoding(factor=100, src_type=np.float32),
pdbx.DeltaEncoding(),
# Encode the difference into two bytes
pdbx.IntegerPackingEncoding(byte_count=2, is_unsigned=False),
pdbx.ByteArrayEncoding(),
]
)
category = pdbx.BinaryCIFCategory(columns)
bcif_file = pdbx.BinaryCIFFile(
{"lyosozyme_md": pdbx.BinaryCIFBlock({"coord": category})}
)
file = NamedTemporaryFile("wb", suffix=".bcif")
bcif_file.write(file)
file.flush()
xtc_size = os.path.getsize(xtc_file_path)
bcif_size = os.path.getsize(file.name)
file.close()
figure = plt.figure()
ax = figure.add_subplot(111)
ax.bar(
[1,2], [xtc_size/1e+6, bcif_size/1e+6], width=0.3,
color=[biotite.colors["dimgreen"], biotite.colors["dimorange"]],
linewidth=0
)
ax.set_xticks([1,2])
ax.set_xticklabels(["XTC", "BinaryCIF"])
ax.set_xlim(0.5, 2.5)
ax.set_ylim(0, 40)
ax.yaxis.grid(True)
ax.set_ylabel("File size (MB)")
figure.tight_layout()
plt.show()
