


biotite.structure.filter_amino_acids¶
- biotite.structure.filter_amino_acids(array)[source]¶
Filter all atoms of one array that belong to amino acid residues.
- Parameters
- arrayAtomArray or AtomArrayStack
The array to be filtered.
- Returns
- filterndarray, dtype=bool
This array is True for all indices in array, where the atom belongs to an amino acid residue.
Notes
Amino acids are identified according to the PDB chemical component dictionary. A residue is considered an amino acid if it its
_chem_comp.typeproperty has one of the following values (case insensitive):D-BETA-PEPTIDE,C-GAMMA LINKING,D-GAMMA-PEPTIDE,C-DELTA LINKING,D-PEPTIDE LINKING,D-PEPTIDE NH3 AMINO TERMINUS,L-BETA-PEPTIDE, C-GAMMA LINKING,L-GAMMA-PEPTIDE, C-DELTA LINKING,L-PEPTIDE COOH CARBOXY TERMINUS,L-PEPTIDE LINKING,L-PEPTIDE NH3 AMINO TERMINUS,PEPTIDE LINKING
Gallery¶
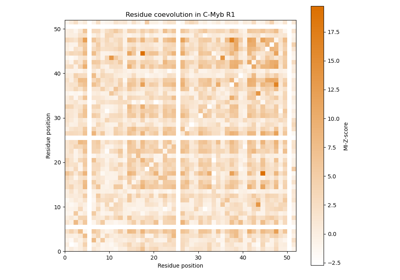
Mutual information as measure for coevolution of residues


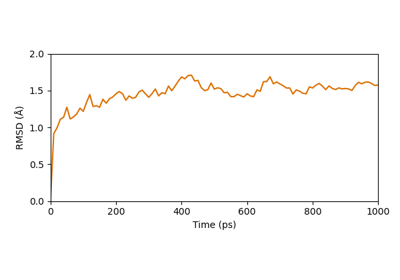

Visualization of normal modes from an elastic network model
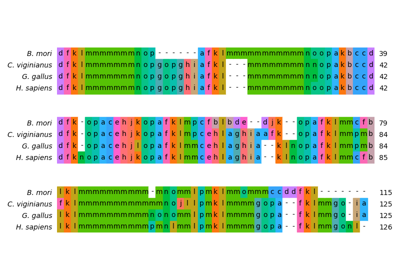
Structural alignment of lysozyme variants using ‘Protein Blocks’


Three ways to get the secondary structure of a protein

