


Note
Go to the end to download the full example code
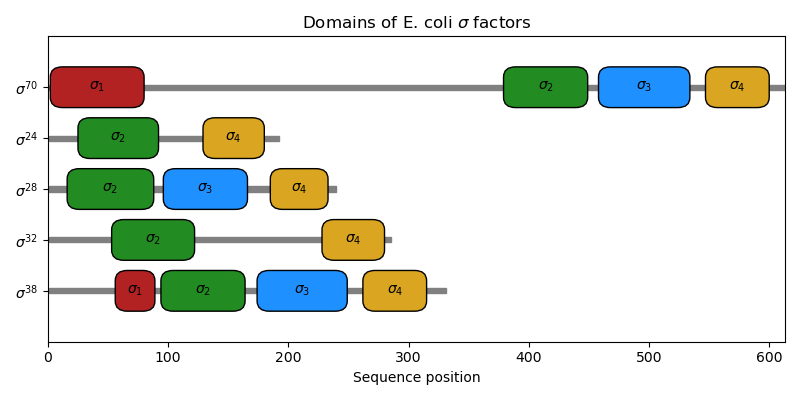
Domains of bacterial sigma factors¶
This script displays the 4 fundamental domains of the E. coli \(\sigma^{70}\)-like \(\sigma\) factors.
# Code source: Patrick Kunzmann
# License: BSD 3 clause
import re
from collections import OrderedDict
import numpy as np
import matplotlib.pyplot as plt
from matplotlib.patches import Rectangle, FancyBboxPatch
import biotite.sequence as seq
import biotite.sequence.io.genbank as gb
import biotite.database.entrez as entrez
# The names of the sigma factors and the corresponding genes
genes = OrderedDict({
r"$\sigma^{70}$": "rpoD",
r"$\sigma^{24}$": "rpoE",
r"$\sigma^{28}$": "rpoF",
r"$\sigma^{32}$": "rpoH",
r"$\sigma^{38}$": "rpoS",
})
# Find SwissProt entries for these genes in NCBI Entrez protein database
uids = []
for name, gene in genes.items():
query = entrez.SimpleQuery(gene, "Gene Name") \
& entrez.SimpleQuery("srcdb_swiss-prot", "Properties") \
& entrez.SimpleQuery("Escherichia coli K-12", "Organism")
ids = entrez.search(query, "protein")
# Only one entry per gene in E. coli K-12 is expected
assert len(ids) == 1
uids += ids
# Download corresponding GenBank files as single, merged file
file = entrez.fetch_single_file(
uids, None, "protein", ret_type="gb"
)
# Array that will hold for each of the genes and each of the 4 domains
# the first and last position
# The array is initally filled with -1, as the value -1 will indicate
# that the domain does not exist in the sigma factor
domain_pos = np.full((len(genes), 4, 2), -1, dtype=int)
# Array that will hold the total sequence length of each sigma factor
seq_lengths = np.zeros(len(genes), dtype=int)
# Read the merged file containing multiple GenBank entries
multi_file = gb.MultiFile.read(file)
# Iterate over each GenBank entry
for i, gb_file in enumerate(multi_file):
_, length, _, _, _, _ = gb.get_locus(gb_file)
seq_lengths[i] = length
annotation = gb.get_annotation(gb_file)
# Find features, that represent a sigma factor domain
for feature in annotation:
if feature.key == "Region" and "note" in feature.qual \
and "Sigma-70 factor domain" in feature.qual["note"]:
# Extract the domain number
# and decrement for 0-based indexing
#
# e.g. 'Sigma-70 factor domain-2.' => 1
# ^
domain_index = int(re.findall(
"(?<=Sigma-70 factor domain-)\d+",
feature.qual["note"]
)[0]) -1
# Expect a single contiguous location of the domain
assert len(feature.locs) == 1
loc = list(feature.locs)[0]
# Store first and last position of the domain
domain_pos[i, domain_index, :] = [loc.first, loc.last]
fig = plt.figure(figsize=(8.0, 4.0))
ax = fig.gca()
# The color for each one of the four domains
colors = ["firebrick", "forestgreen", "dodgerblue", "goldenrod"]
# Draw each sequence
for i, (gene_name, domain_pos_for_gene, length) \
in enumerate(zip(genes.keys(), domain_pos, seq_lengths)):
# Add base line representing the sequence itself
ax.add_patch(Rectangle(
(1, i-0.05), length, 0.1, color="gray"
))
# Draw each domain
for j, ((first, last), color) \
in enumerate(zip(domain_pos_for_gene, colors)):
if first != -1 and last != -1:
# FancyBboxPatch to get rounded corners in rectangle
ax.add_patch(FancyBboxPatch(
(first, i-0.4), last-first, 0.8, #color=color,
boxstyle="round,pad=0,rounding_size=10",
ec="black", fc=color,
mutation_aspect=0.02
))
ax.text(
x=(last+first)/2, y=i, s=fr"$\sigma_{j+1}$",
ha="center", va="center"
)
ax.set_xlim(0, max(seq_lengths))
ax.set_xlabel("Sequence position")
# Inverted y-axis
ax.set_yticks(np.arange(len(genes)))
ax.set_yticklabels(list(genes.keys()))
ax.set_ylim(len(genes), -1)
ax.set_title(r"Domains of E. coli $\sigma$ factors")
fig.tight_layout()
plt.show()
